catcheR_step1QC - Pooled Cloning Step 1 QC¶
This step complements Supplemental Protocol 1.
In a new working folder, prepare the following files:
Fastq/fq or fastq.gz files with demultiplexed read 1 from the NGS run.
A CSV file with the shRNA names and their full sequences:
SMAD2.1,GCAAGTACTCCTTGCTGGATTGCTCGAGCAATCCAGCAAGGAGTACTTG ...
(Optional) A .txt file with a newline-separated list of clones of interest in the format BC_UCI (e.g., from a subsequent iPS2seq-edited-hPSC experiment). Each clone is identified by its shRNA barcode and UCI separated by an underscore:
CAAGAGCC_CATCGT ...
Run
catcheR_step1QC:catcheR_step1QC( group = c("docker", "sudo"), folder, fastq.read1, DIs = 100, ratio = 10, plot.threshold = 2000, clones = NULL )
`catcheR_step1QC` arguments:
group: string, either “sudo” or “docker” depending on your user group. (For Docker group setup, see: https://docs.docker.com/engine/install/linux-postinstall/ *)folder: string with the path to the working directory.fastq.read1: string with the filename from step 1a.DIs: integer, minimum number of diversity indexes (DIs, pseudo-unique reads) for the most represented shRNA matched to a given UCI-BC. Used withratioto select reliable UCI-BC/shRNA associations. (Default = 100)ratio: integer, the minimum ratio between the DIs of the most represented and second most represented shRNAs. Used withDIsto confirm assignment. (Default = 10)plot.threshold: integer, minimum number of DIs per UCI-BC to be included in bar plots.clones: (optional) string with the .txt file name from step 1c.
Example usage:
catcheR_step1QC( group = "docker", folder = "path/to/folder", fastq.read1 = "filename.fq", clones = "filename.txt" )
`catcheR_step1QC` key outputs:
reliable_clones_swaps.csv— lists UCI-BCs with strong evidence of shRNA-barcode swap. Can be used as input for SPFourEight.Bar chart showing the number of DIs for each clone above the
plot.threshold.Bar charts showing the number of DIs associated with each barcode, shRNA, and reliable swap.
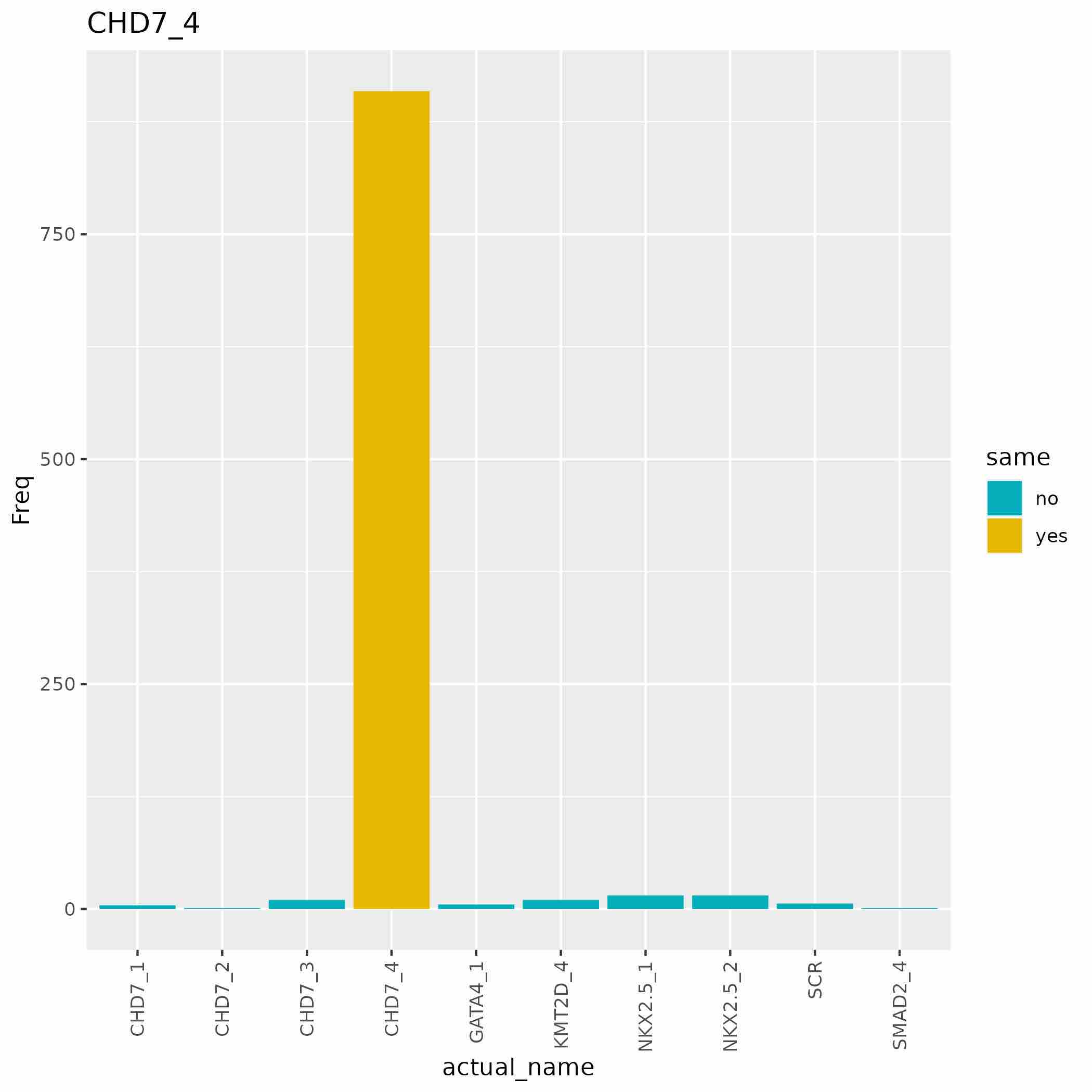
(If the “clones”” argument is provided)
CSV files and bar charts showing the number of DIs for each shRNA matched to each clone of interest (see FigureSPFourTwo).